Introduction#
This pipeline is designed for the retrieval, processing, subsampling, and phylogenetic analysis of the latest dengue virus sequences. It leverages Snakemake, a powerful workflow management system, to ensure reproducibility and efficiency in bioinformatics analyses.
Pipeline#
Here is an overview of the pipeline that we will construct. For simplicity (and due to the extended runtime of some software) we will implement the main pipeline in this tutorial, disregarding the BEAST branch.
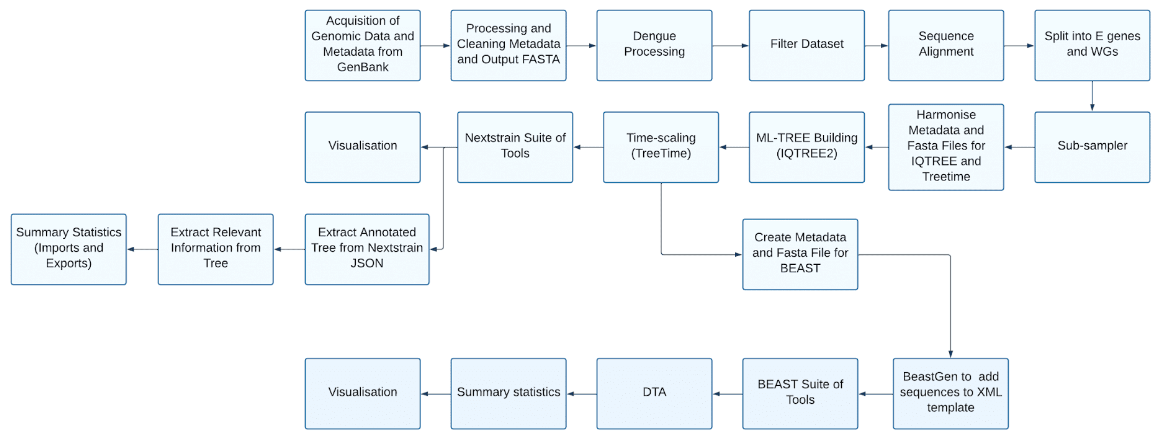
Step by step of the pipeline#
The steps in the pipeline are as follows (see the Dengue repository for more details):
Acquisition of Genomic Data and Metadata from GenBank
Clean metadata and FASTA files
Filter for sequences from SEA
Split into serotype, add serotypes to sequence name and generate sequence specific metadata
(Future step not currently implemented) Verifying Serotypes and Genotypes
Sequence alignment
Segregating E gene and Whole Genomes and performing quality control
Subsampler
Correct metadata and fasta files into the correct format for iqtree and treetime
ML-Treebuilding
Build time-calibrated trees
Infer “ancestral” mutations across the tree (nextstrain)
Translate sequences (nextstrain)
Discrete trait reconstruction (nextstrain)
Export for visualisation in Auspice (nextstrain)
Extract annotated tree from nextstrain JSON format
Extract information from tree
Quantify number of exports and imports from desired country